Gene regulations and 3D structure
생물체 유전 정보의 총체인 유전체는 진핵생물의 핵 내에서 DNA 분자가 꼬이고 정리된 상태의 입체구조를 이룬 상태로 존재한다. 유전체가 가지는 입제 구조를 통해 유전체 상의 특정 지역끼리의 상호작용, 단백질을 매개로 하는 상호작용 들이 발생하고, 또 새로이 유도된다. 이러한 입체구조로 발생하는 고차원의 유전체 상호작용은 두 개 이상의 지역이 관여할 수 있고, 그 중 enhancer - promoter 간의 상호작용은 on-off 보다 양적으로 수치화가 가능하다고 알려져 있다. 이러한 DNA 입체 구조는 생물체 내 조직, 기관에 따라 그 패턴이 상이하며 각기 다른 조직/기관에서의 유전자 발현을 유도한다. 따라서 단일 세포 수준에서의 DNA 입체구조를 밝히는 작업에 대한 필요성이 대두되었다.
Calculation of genome 3D-structure
최근 널리 사용되고 있는 DNA 입체 구조를 밝히는 방식은 Hi-C sequencing 방식을 통한 것이다. 아래와 같은 방식으로 통해 Hi-C sequencing read 생성이 가능하다.
1. 채취한 샘플에 대해 제한효소 (Hind3)를 처리하여 해당 제한효소가 인식할 수 있는 DNA상의 인식 서열을 절단한다.
2. 절단된 DNA 분자 말단에 빈 상보적 염기 서열을 biotin 이라는 인식 단백질을 달고있는 염기를 사용하여 메꿔주고
3. 인접하게 존재하는 잘려있는 DNA말단이 연결될 수 있도록 Ligase 효소 처리를 해 준다.
4. 시퀀싱을 위한 단편화 작업을 수행 후
5. biotin 단백질이 붙어있는 복합체만을 인식해 (interaction 이 발생한 부위의 두 DNA 가닥이 붙어있는 복합체) 분리하고
6. 시퀀싱을 수행한다.
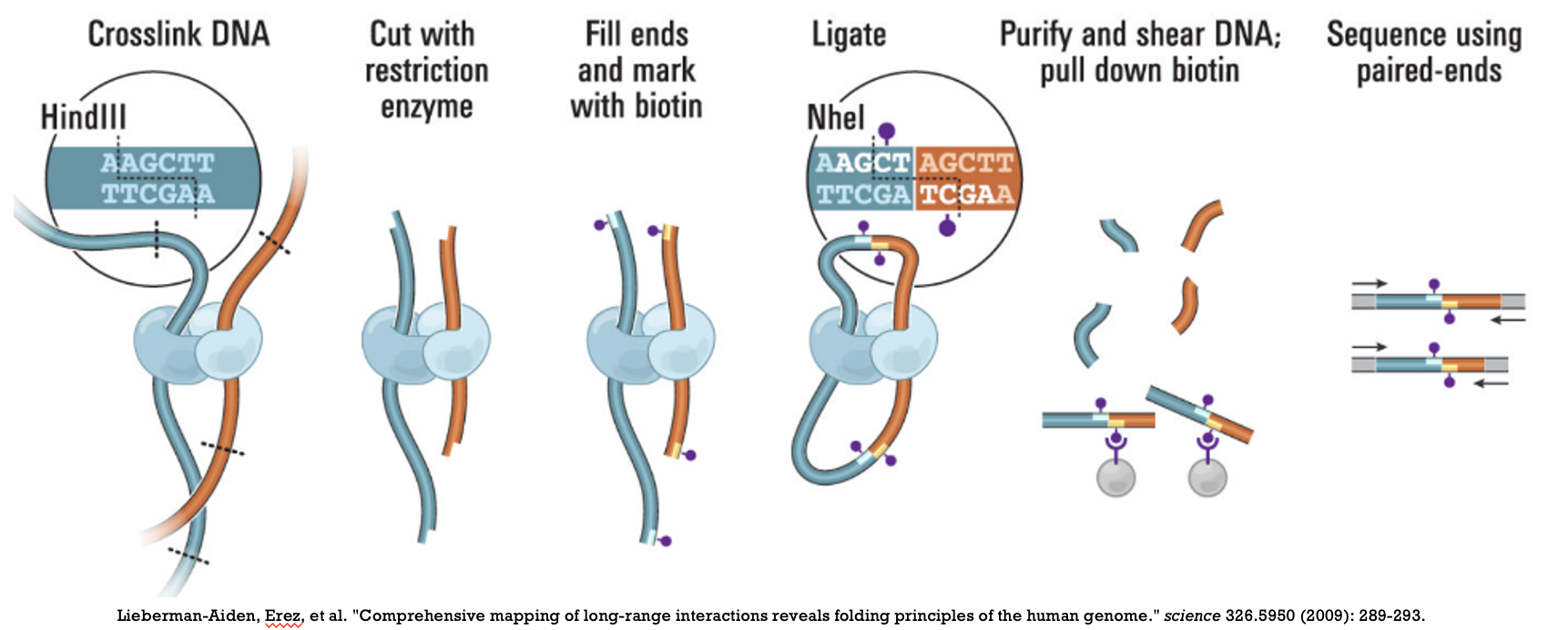
위와 같은 방식으로 Hi-C 시퀀싱을 수행하면 DNA 상에서 상호작용하고 있는 지역과 인접한 두 지역에 대한 한 쌍의 염기서열 정보를 획득할 수 있다. 그 데이터를 활용하면 다음과 같은 contact map 정보로 변환할 수 있다.

최근에는 또한 시퀀싱 기술의 발달로 단일 세포에 대한 Hi-C 데이터의 생성도 가능해졌다. 그런데, bulk-cell Hi-C 데이터와 달리 단일 세포 (single-cell) Hi-C 데이터의 경우 발견할 수 있는 contact (상호작용) 정보가 너무 적고, 단일세포로부터 추출한 데이터로서 가지는 데이터의 희소성, 노이즈가 크게 작용해 실제 해당 세포 상에서의 입체 구조를 특정하기 어렵다는 단점이 있다. 따라서 이러한 단점을 보완하기 위한 데이터 처리 측면에서의 다양한 방법론이 개발되어오고 있다.
Si-C method
본 논문에서는 위에서 언급된 단일세포 Hi-C 데이터의 해석 및 처리에 대한 다양한 문제점을 극복하기 위해 Si-C (Single-cell chromosome conformation calculator) 방법론을 소개한다. Si-C는 베이지안 이론을 기반으로 개발되었으며, 완전한 데이터 기반의 모델링 프로세스로 통계적 추론을 수행한다. Si-C는 최종적으로 DNA의 입체 구조를 예측하는 작업을 수행하고, 이 과정은 베이지안 모델 상에서 주어진 Hi-C contact constraint (C, 데이터 상에서의 contact 정보)가 만족될 때, 그 확률이 최대가 되는 DNA 입체 구조 (R, 3D genome conformation)을 찾는 과정으로 이루어진다.
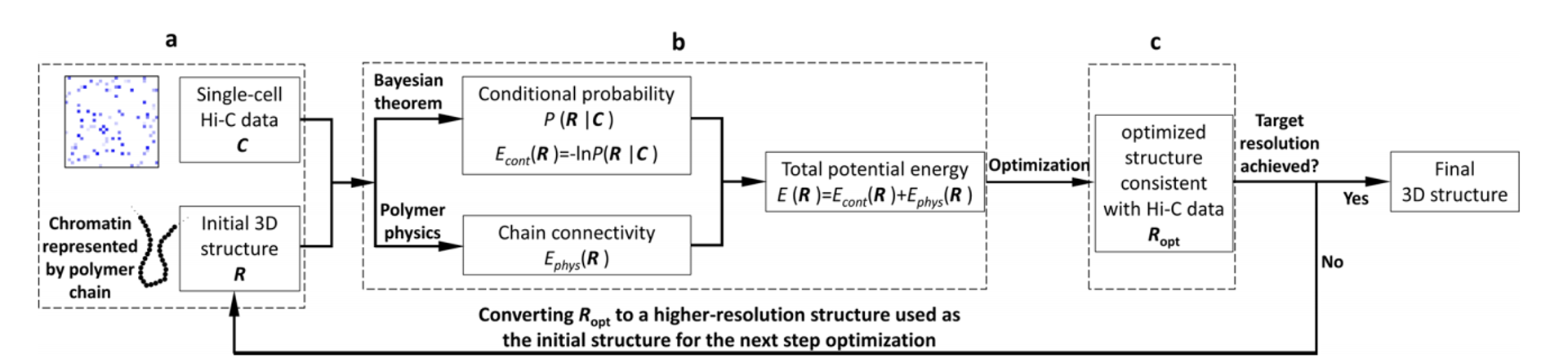
이 과정에서 저자들은 3D genome conformation (R), Single-cell Hi-C data (C) 두 경우에 대해 조건부확률 (P(R|C))을 정의하였다. 물리적 최적치 (E_phys(R))와, 음의 자연로그를 취한 조건부확률 (E_cont(R))의 합인 E(R)을 최소화할 수 있도록 최적화를 수행하게 했다. 계층 최적화 기법을 도입하여 최적화 과정을 보다 효율적으로 수행할 수 있게 했다. Si-C 방법론은 기존에 개발 된 두 가지 방법론 (NucDynamics, ISD)의 장점을 결합하여 생물학적으로, 또한 통계적으로 모두 유효한 DNA 입체구조에 대한 추론을 가능하게 했다.
본 방법론은 크게 세 가지 단계로 구분되고, 그 단계는 다음과 같다.
1. 염색체 모델링 (DNA 시퀀스의 polymer chain 형태로의 표현을 통한 입체구조 형태 초기화 작업과, Single-cell Hi-C 데이터 준비)
2. 염색체의 입체 구조에 대한 총 위치 에너지 정의
3. 총위치에너지의 최적화를 통한 최종 입체구조 결과 도출
Results
저자들은 Si-C 방법론을 활용한 데이터 분석을 통해 해당 방법론의 유효성을 검증하고, 또한 이 방법론의 효용성을 추가로 제시하였다.
The percentage of contact restraints that are violated in the 3D model

본 모델이 sincle-cell Hi-C 방식으로 생산된 시퀀싱 데이터로부터 얻을 수 있는 contact 정보를 수집하므로 본 방법론이 생물학적으로 유효한 입체구조를 탐지해낸다는 것을 증명하기 위해서는 이 모델이 데이터를 기반으로 예측한 결과와 실제 실험적으로 측정한 DNA 상에서의 입체구조 - contact 정보과 일관되는지에 대한 확인이 필수적이다. 따라서 저자들은 두 개의 bead (본 모델에서 DNA를 polymer로 표현하기 위해 차용한 구조)가 두 개 이상의 bead 직경 거리 이상으로 떨어져 있는 경우 Violation 이라고 정의하고, 예측 결과 내에 Violation의 발생 비율을 확인하였다. Figure a. 에서 기존의 방법론인 NucDynamics에 비해 Si-C의 예측이 보다 낮은 수준의 violation percentage를 보이는 것을 확인할 수 있었고, 같은 Si-C 방법론 상에서는 보다 높은 해상도에서 좋은 성능을 보이는 것으로 확인되었다. Figure b. 에서는 예측된 입체 구조의 공간 상 거리의 중간값과 FISH를 통해 측정한 공산 상 거리의 중간값이 8개 세포에 대해 구한 뒤, 이들의 선형성을 확인하였다.
Agreement between the experimental and back-calculated data
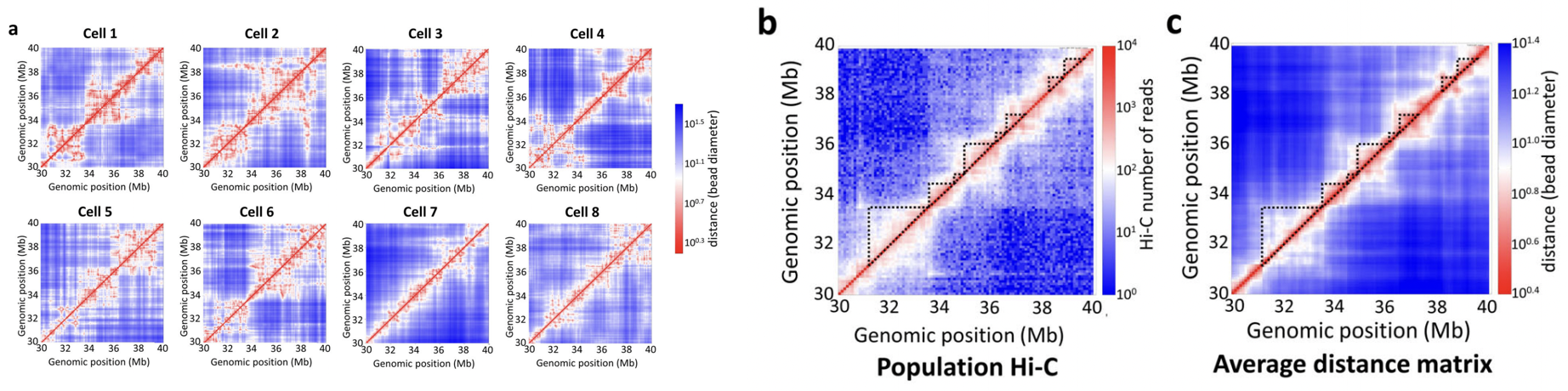
저자들은 또한 입체구조 예측과 Population Hi-C 데이터로 확인할 수 있는 입체구조가 일치하는지 여부를 확인하고자 하였다. Figure a 의 경우, 개별 세포에 대한 예측된 입체구조 데이터에 대한 시각화 결과물이고, 이로부터 서로 다른 세포들은 서로 상이한 패턴의 DNA 입체구조를 가진다는 것을 유추할 수 있다. 개별 8개 세포에 대해 예측한 입체구조 데이터의 평균을 구한 것이 Figure c 이다. 이는 전체 세포의 Hi-C 데이터를 한 데 모아 Population 수준의 Hi-C 데이터를 활용해 만든 시각화 결과물 (Figure b)과 유사한 것을 확인할 수 있다.

위 그림은 예측된 개별 세포에서의 입체 구조 데이터로부터 추론 가능한 TAD의 경계 (Figure f, peak)가 실제 Population Hi-C 데이터상에서 유추되는 것과 유사함을 보여줌으로써 Si-C 방법론의 유효성을 검증한다.
Reveals conserved 3D whole-genome architecture in all cells
본 연구에서는 또한 예측된 개별 세포의 입체 구조에 실험적으로 밝혀진 유전체 상의 특정 지역들에 대한 주석 처리 및 시각화를 통해 예측 결과가 생물학적으로 타당함을 추가로 검증하였다. 또한 본 연구에서 생성한 결과로부터 관찰할 수 있는 유전체 입체구조의 특징들은 실험적으로 검증된 이전 연구의 결과들과도 일관됨을 보이고 있다.

10kb-resolutions gives more details
다음 그림은 단일 세포에 대한 입체구조 예측을 통해 개별 세포에서 어떤 형태의 염색질 상호작용이 발생하는지 유추할 수 있게 되었음을 시사한다. 보다 높은 해상도로 예측을 수행할 수록 염색질이 Loop, Hairpin 등의 특이 구조를 이루고 있는지 여부를 확인할 수 있다.

Discussion
- 본 연구에서는 베이지안 확률을 활용, 단일세포 Hi-C 데이터를 입력으로 받아 유전체 입체구조를 예측하는 방법론을 개발하였다. Si-C 내부에서는 시퀀스 데이터로부터 수집한 Contact 데이터와, polymer 형태를 차용한 에너지 항을 활용하여 유전체의 입체구조를 예측한다.
- 방법론의 측면에서는 주어진 데이터 대비 최대의 확률을 가지는 입체구조를 판단하기 위해 조건부확률을 정의하였고, 경사하강 알고리즘을 채택하였다.
- 다양한 방식으로의 검증을 통해 본 방법론이 높은 해상도에 대해 신뢰할 수 있는 단일 세포 데이터에 대한 유전체 입체구조를 예측해 낼 수 있음을 확인하였다.



